���w | ���w
����(n��i)�Ҍ����B�iˎ������(sh��)ʮλ��(zh��)�I(y��)ˎ���ṩ��ˎָ��(d��o)��Ͷ�V�ᾀ:(020)66843700
ˎ����B | ˎƷ���} | ˎƷ��(d��o)ُ | �����l�� | ����(w��)ָ��
ˎ���ӑB(t��i) | ������ˎ | �t(y��)Ժ�l�� | ˎ������ | ������ˎ
ˎ���ӑB(t��i) | ������ˎ | �t(y��)Ժ�l�� | ˎ������ | ������ˎ

�༪�����} >> �༪���R��ǰ����ˎЧ�W(xu��)�о�
�༪���R��ǰ����ˎЧ�W(xu��)�о�
- ��Դ�� �ٝ�����ˎ���W(w��ng) �l(f��)���r�g��2007-3-13 14:11:00
��Ҫ��B
Sorafenib������ڌ�CRAF��ø���Ƅ��Ȍ�(d��o)���M�нY(ji��)��(g��u)-�����u�r�����������б��l(f��)�F(xi��n)�ġ����w�⣬sorafenib��CRAF��Ұ���ͼ�ͻ׃��(V600E)BRAF����Ч�����Ƅ���Sorafenib߀���ƅ��c�[���Mչ��Ѫ�����ɵĎNRTKs��������VEGFR-2��С��VEGFR-2��VEGFR-3��PDGFR-�£�F(xi��n)LT3��c-KIT���������ٰ�����ɫ���������ٰ����Y(ji��)�c����(x��)��ϵ�У�sorafenib�����˼�(x��)����(n��i)MAPK;���Ļ��A(ch��)���ữˮƽ���������_ͻ׃KRAS��NSCLC��(x��)��ϵNCI-H460��A549��(x��)���o�����á��ڱ��_Ұ����ͻ׃��KRAS��BRAF�������ٰ������ٰ�����ɫ�����ͽY(ji��)�c����(x��)��ϵ�У�sorafenib�����˼�(x��)����(n��i)MAPK;������̖����(d��o)��
Ҫ�c��
��Sorafenib��RAF��ø�ď����Ƅ��������Ƶ�RAF��ø����CRAF��Ұ���ͺ�ͻ׃��(V600E)BRAF�����[���Mչ�аl(f��)�]���õĎNRTKs��
�� Sorafenib���w��(n��i)���ЏV�V�Ŀ��[�����ԡ��ڱ��_ͻ׃RAS��/��BRAF��ؓ(f��)��С��ģ�ͣ�����(j��ng)RAS/RAF/MEK;������(d��o)���L��̖��ؓ(f��)��С��ģ���У�sorafenib���ί��������[�������L��
��Sorafenibͨ�^����VEGFR2��VEGFR3��PDGFR-�£����p����Ѫ��������̖�Ă���(d��o)���������ͨ�^ֱ�Ӻ��g�әC�ƹ�ͬ�����[�����L��
�w���о�
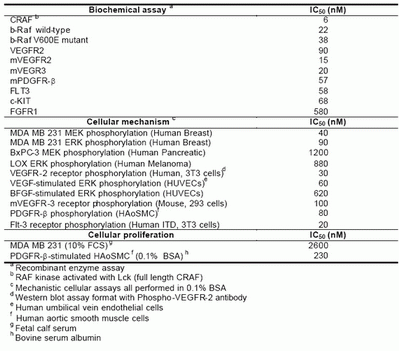
��4-1���Y(ji��)��sorafenib���������Լ���(x��)��ˎ��W(xu��)�Y(ji��)��(21)��sorafenib����CARF��Ұ����ͻ׃(V600E)BRAF�ď����Ƅ����w��IC 50�քe��2nM��22nM��38nM(22)��sorafenib߀���ƎN���c�[���ݻ���Ѫ�����ɵ�RTKs������FLT3�� c-KIT��VEGFR-2��VEGFR-3��PDGFR-�¡���ʹˎ���ȸ��_10��M��sorafenib��MEK-1, ERK-1, EGFR, HER2/NEU, c-MET, PKA, PKB, IGFR-1, Cdk-1/cyclin B, PIM-1, GSK 3-, CK-2, PKC-, PKC-ß, �� PKC-��
Sorafenib��ţp59 FYN��ø��IC50��780nM���f��ˎ��˼�ø�]���@�����������á����Ǽ�ø��λ��sorafenib������A3����Ͱ�D1�Ͷ�ަ�AM3���w���p�ĵ��������ã�IC50�քe��1.6��M��2.0��M��3.1��M��
��4-1��Sorafenib����ø��λ���w�����
���w�⣬sorafenib�����ٰ������ٰ�����ɫ�����ͽY(ji��)�c����(x��)����RAF/MEK/ERK;������̖����(d��o)���@�����������ã����F(xi��n)����_Ұ���ͻ�ͻ׃��KRAS��BRAF�ļ�(x��)����pERKˮƽ���͡�Ȼ��������NSCLC��(x��)��ϵA(ch��)549��H460��(x��)������ʹsorafenib�ĝ�ȸ��_20M����Ȼ�]���^�쵽ˎ�RAF/MEK/ERK;������̖����(d��o)���������á�ˎ��@Щ��(x��)��ϵ��(n��i)ERK���ữ�o�������õ�ԭ���в������Sorafenib�����_��������Ѫ�܃�(n��i)Ƥ��(x��)����(n��i)��(VEGFR2)���ܰ����Ƀ�(n��i)Ƥ��(x��)����(n��i)��(VEGFR3)�����˴���}ƽ������(x��)����(PDGFR-��)�����w�����ữҲ���������á�VEGF��PDGF���w���[����Ѫ������������Ҫ����(15, 20)����ˣ�sorafenibһ����ͨ�^����VEGFs��PDGF���w������[����Ѫ�����ɣ���һ����ͨ�^����RAF/MEK/ERK;������̖����(d��o)�������[�����L��
�w��(n��i)�о�
.1).�΄�ˎЧ
�ڱ��_ͻ׃��KRAS(HCT-116, DLD-1, Mia-PaCa-2)��BRAF(Colo-205��HT-29)���[��ģ���У�sorafenib�@ʾ�˿��[�����á����ڱ��_�����ɷNͻ׃�����MDA-MB-231ģ�ͣ�ˎ����Ȼ��Ч�������_Ұ����RAS��BRAF�������^���_EGF��HER2���L�������w�����ѳ�����(x��)����sorafenibҲ�@�F(xi��n)�˯�Ч���@Щ���wҲ��ͨ�^RAF/MEK/ERK;������(d��o)��̖��Sorafenib���@Щ�[��ģ�����@�F(xi��n)����Ч����ʾRAF��ø���Ƅ����H�����_RAS��/��ͻ׃��b-Raf�����[����Ч��������ͨ�^����̖����(d��o)ͨ·���f��̖�ġ����L�������w�^�ȱ��_�ĵ��[��Ҳ��Ч�����sorafenib�����_���ͻ׃��FLT-3��MV4��11AMLģ��Ҳ��Ч����4-2���Y(ji��)��Sorafenib�����[�����w��ֲ��ģ�����@ʾ���w��(n��i)�V���Ŀ��[�����ԡ�
��4-2�����R��ǰ���N��ֲ��ģ���У�sorafenib�Ŀ��[������
������7.5mg/kg/�쵽90mg/kg/��r��sorafenib��RENCAС��RCCģ����Ҳ�@ʾ�˺��Ŀ��[������(�[�����L�����ʞ�30%��84%)��
�ڟo����С��A498��CAKI-1���I�����N��ֲģ���У��M����ˎ��ij����Ŀ��[����Ч�о����ڄ������_60mg/kg�r��sorafenib��Ȼ�]���@������CAKI-1�[�������L(25)��Ȼ������(d��ng)sorafenib�Ą�����60mg/kg�r�����о����g��70%��A498�[���ĵĴ�С�]�мӱ�(26)�������ڌ��սM�[�������L�̶ȣ����о���ˎ��]���@�F(xi��n)�����@���[�����L�������á������������߽oˎ�r�g���L��sorafenib���ܕ����F(xi��n)�����@���[�����L�������á��ڃɷN�I��ģ���У�����(x��)����Sorafenib�������Բ�ͬ��ˎ������Ҳ�в�e������ԭ���в������
��DLD-1�Y(ji��)�c��ģ�ͣ����sorafenib�ί�����Ч��(27)���ί���Sorafenib�ί���ˎ�С�[������[�������L�����������á�ˎ��[�����L�����t�����c�ί����m(x��)�r�g�����ȣ����c�ί������[���Ĵ�С�o�P(gu��n)���c��һ���̵į�Ч��ȣ��ɯ��̡�ÿ����10����ί�ʹ�[�����L���t����2������30��ij��m(x��)�ί�ʹ�[�����L���t����3����ֵ��ע����ǣ��ڵ�һ�����ί����[�����L�����ƣ�������ֹ�ί����[�����LҲ�֏�(f��)�ˣ��@�r�_ʼsorafenib�ڶ����̵��ί���Ȼ�����_�������[�����L��Ŀ�ġ�
.2).���ÙC��
�ڶ�N���w�[��ģ�����о���sorafenib�����ÙC��������HT-29, DLD-1, HCT-116, �� Colo-205 �Y(ji��)�c��ģ��, MDA-MB-231�鰩ģ��ģ��(21, 28)����ÿ���о��У�ؓ(f��)�ɴ�s200�����[���Ą���o���ͺ�Mia-PaCa-2����sorafenib(30-60mg/kg/��)������5�졣ĩ�νoˎ��3С�rȡ���[������WesternȾɫ��/�����߽M��(IHC)�������u�rsorafenib��RAF/MEK/ERK��̖����(d��o)ͨ·�����á���һЩ����У��[����Ʒ߀�M���˿�CD31��ɽ����¡���wȾɫ��Ӱ�����Ѫ���ܶ����u�rˎ�����Ѫ�����ɵ��������á���HT-29��Colo-205�[��ģ�͵õ��ĽY(ji��)����C���@Щ�u�r�ó��ĽY(ji��)Փ��
ÿ�M4ֻС����ֲ���˽Y(ji��)�c�[������(d��ng)�[���L����100��200���˕r��С��ڷ�������30mg/kg��60mg/kg��sorafenib��ÿ��1�Σ���5�졣�ÿ�-pERK�Ϳ�-ERK���w�M��Western blot�������Y(ji��)��������sorafenib������HT-29��(n��i)RAF/MEK/ERK��̖�D(zhu��n)��(d��o)ͨ·�Ļ�����nj�Colo-205�˽Y(ji��)�c���oЧ���mȻ��HT-29��Colo-205�Y(ji��)�c���[�����N��ֲģ���У�sorafenib���@���������[�������L��������Western blotting��IHC�ķ����քe����Colo-205�[������δ�ܰl(f��)�F(xi��n)ˎ�ERK1/2���ữ���@�����������á���ʾsorafenib���ί�δ����Ч���RAF/MEK/ERK;������̖����(d��o)����Colo-205��HT-29��(x��)����(n��i)������ͬ�ӵ�V600E b-Rafͻ׃��������@Щ��(x��)��������ֳ��ԭ���ܴ�������һ�l;����������RAF/MEK/ERK��(li��n)����(y��ng)�����Colo-205�[����(n��i)��ERK1/2���c���෴���M����Colo-205�[���ӱ��У�sorafenib���ί�δʹRAF/MEK/ERK;������̖����(d��o)�ܵ����ƣ�����Colo-205�[���ӱ��cHT-29�[���ӱ�һ�ӣ�Ѫ�܅^(q��)��e���ܶȶ�������50%��80%��
���x���ˎ��W(xu��)
�ϳ���sorafenib�����N���x��(M-2, M-4��M-5)�����о���������Ұ����c-Raf��b-Raf��mPDGFR-��, mVEGFR-2����FLT��3����������(38)��M-2��M-5��mPDGFR-�µ������������@����sorafenib��Ȼ����sorafenib��c-Raf��Ұ���ͼ�ͻ׃��b-Raf�����������@������M-2��M-5��Sorafenib��FLT-3�������������@����M-5��M-4��
���ā��f�����x��ļ�(x��)����(n��i)Ч��(y��ng)�csorafenib�Ļ���һ�¡�ERK���ữˮƽ�����ں���RAF/MEK/ERK;����̖����(d��o)���Ƴ̶ȣ���MDA-MB-231���ٰ���(x��)����(n��i)���@Щ�����ERK���ữ���������Û]�в�e��IC50��90��151nM���ɂ����x�M-2��M-5����PDGF�錧(d��o)���˴���}ƽ������(x��)�����������������@������sorafenib��NIH-3T3�w�S��(x��)���D(zhu��n)ȾС��VEGFR2���^��ˎ�VEGFR2�Ԅ����ữ�������õ�IC50�����x���IC50��12��30nM���csorafenib��ț]���@����e��
BAY 67-3472(���x��M-2)�Ŀ��[������
�ڃɂ��������о��У���MDA-MB-231�����ٰ�NCr����ģ�ͣ����^��BAY67-3472(M-2)����ĸ�w������Sorafenib�Ŀ��[��Ч��(y��ng)(39)�����@�ɂ�ԇ��У�ÿ���Ƅ��Ŀڷ�������30, 60��120mg/kg���oˎ������QD��9����ǰ���������ñ��о��еĽoˎ������sorafenib�ڷ�������30mg/kg�r������ȫ����MDA-MB-231�[�������L����ˣ������@һ����ֱ�ӱ��^�ɂ�������Ŀ��[�����ԡ����⣬������ǰ�Ľ�(j��ng)����(d��ng)�������^120mg/kg�r��sorafenib��ȫ��ˎ���Ȳ������ߣ�����ڃɂ��о���BAY67-3742���õ���߄�����60��120mg/kg��BAY67-3742 120mg/kg�Ą������ѽ�(j��ng)�_�����о���ʹ�õ��x�΄�PEG400/MSA�ܽ�����O�ޡ�
Sorafenib�M�������������ã��w�H����4%-5%���]�Є�����������ǰ�������ڴ�ģ����Sorafenib�į�Ч�@�����cģ�͌��սM��ȣ��[�����L������(TGI)��80%-91%��BAY67-3472��������Ҳ�^�ã������w�p�p��2%-7%���]�Є���������BAY67-3742������30mg/kg�r��TGIs��38%��������120mg/kg�r��TGIs��68%����ˣ�BAY67-3472�����Ï��Ⱥ���Ч�Զ�����sorafenib����ˎ�oˎ;�����oˎ������ͬ�ėl���£�BAY67-3472�Ą�����120mg/kg�r�a(ch��n)�����[�����L��������(TGI��63%��68%)������sorafenib������30mg/kg�r�a(ch��n)����Ч��(y��ng)(TGI��80%��91%)��
�چΪ����о��Мy����NCrС��ڷ�BAY67-3472��BAY67-3472��sorafenib��ˎ�������W(xu��)����(sh��)��BAY67-3472��Ч�^���cNCrС��ڷ�BAY67-3472��õ���Ѫˎ����^�����P(gu��n)��BAY67-3472�ڷ�������30mg/kg�r���õ���ˎ��Ѫ�{�_����(Cmax)������4mg/L�� ˎ�r��������e(AUC)��25mg*h/L����ͬ�ӄ�����sorafenib�õ���Cmax��BAY67-3472��10������33mg/L��AUC��326 mg*h/L(40)������BAY67-3472��һ���ֿ��[����Ч��ͨ�^�����w��(n��i)�D(zhu��n)������ĸ�wˎ��sorafenib�����F(xi��n)�ġ��ڷ�������30mg/kg ��BAY67-3472��Ѫ�{sorafenib�ĝ�ȷdz��ͣ�����ˎ��7h���cBAY67-3472��Ѫˎ�����ͬ��Sorafenib�ڷ����ٴη���BAY67-3472���õ���BAY67-3472��AUC(0-24h)�s��58%-71%���ڷ�30��60mg/kg BAY67-3472�õ��ĸ��������Ѫˎ��ȶ����ڄ���������
��֮���@Щ��(sh��)��(j��)�f�����ڷ�sorafenib����Ҫ���x��BAY67-3472��������ˎ��Ŀ��[��Ч��(y��ng)���ڄ������^����У�BAY67-3472�į�Ч�^ĸ�w������͡��ڷ���õ���Ѫ�{ĸ�wˎ�����^�͵������ѽ�(j��ng)�����f���˴��x��Ч��(y��ng)�^����ԭ��Ȼ������Ȼsorafenib��Ҫ���x��BAY67-3472�����о��в�����ȫ�U��BAY67-3472��sorafenib���[�����ԵČ��Hؕ�I��
ӑՓ�ͽY(ji��)Փ
����sorafenib�w��Ч��(y��ng)�����ڼ�(x��)���C���о��Ќ�CRAF���������ã������w��(n��i)�ďV�V�������ã�sorafenib���x����R����ˎ���x֮һ��
Sorafenib�rc-Raf��ͻ׃(V600E) BRAF�ď����Ƅ���߀�������[���ݻ��^��������Ҫ���õĎNRTKs������VEGFR-2��VEGFR-3��PDGFR-��,c-KIT��FLT-3���ڱ��_ͻ׃RAS��/��BRAF�[��ģ�ͣ����������L����ͨ�^ras/raf/MEK;�����f��̖���[��ģ���У���sorafenib�ί��[��ؓ(f��)��С���ʹ�[�����L�ܵ����ơ��������[�����L�������⣬sorafenibͨ�^�����[����Ѫ�����ɣ��ɴ�ͨ�^ֱ�Ӻ��g�әC�ƿ����[�����L���о���ˎ�������w����Ҫ���x��(M-2)���w��(n��i)����Ч��(y��ng)���Y(ji��)���������x�����Ч�Ժ����Ï���С��sorafenib��
TAG�� �༪��
���P(gu��n)ˎƷ

���ˎ�v��ۙ


ʮ��ȫ���B�i����ˎ����ȫ���Ҍ����B�iˎ�������wȫ���^(q��)���Д�(sh��)ʮλ��(zh��)�I(y��)ˎ����
�ٝ������̳ǡ���(li��n)�W(w��ng)ˎƷ������(w��)�Y���C���� ��C20110003


�[���ƌ�(d��o)��
�����T����
���TˎƷ
- �༪��
- ����ɳ
- ���_�P
- ���S͡
- �漪�W
- ��(f��)���f�����z��
- ��(f��)���t��ɼ�z��
- ��(f��)�����z��
- ʳ��ƽɢ
- ��һ�z��
- �������z��
- ����ƽ��
- ��(f��)�������z��
- ���ɢ�Y(ji��)��
- ��κ��Ʀ��
- ���Y�����ڷ�Һ
- �����z��
- �Ώ�(f��)��Ƭ
- ־���z��
- �fđ���Ϳڷ���Һ
- �庣�`����
- �J�S�z��
- �S���_
- ��Ī��
- ���w��
- ������
- ������
- ������̹
- ���Y����Ƭ
- ������
- ������˾
�[����ˎƷ
- ���Ʋ��N������
- �[����
- ���
- ��(j��ng)��
- �����
- Ƥ�w�Բ���
- �ۿ�
- �L(f��ng)�����߿�
- ���XѪ�ܿ�
- ����
- ������ֳ��
- �I�B(y��ng)������
- �t(y��)�����
- ˎƷ�f����������
- �[����
- ���
- ��(j��ng)��
- �����
- Ƥ�w�Բ���
- �ۿ�
- �L(f��ng)�����߿�
- ���XѪ�ܿ�
- ����
- ������ֳ��
- �I�B(y��ng)������
- �t(y��)�����
- Miki��̫�X�Ͻ��p��݆��LS-1
- YX301��ָ�Aʽ�}��Ѫ���x
- �Wķ������w��ӋMC-140
- �~�S݆��H009B
- ����˹������֬�z��
- �վ�����ʽ���Ѫ��ӋWS-720
- ���A�S䓹�݆��HF6-02
- ِ�A٢��Savat������ʽȫ�Ԅ����Ѫ��Ӌ�ռ���
- �~�S����ͨ�õ��_810/820/821/830/831
- �����_������HA-512R
- ���A�S�X�Ͻ�Ҹ�¹�HF7-07S
- �ݰ���ԇ����50Ƭ��
- EW3108�ϱ�ʽ�w�������Ѫ��Ӌ
- �Wķ���ϱ�ʽ���Ѫ��Ӌ HEM-746C
- �Ɓ���ԡ��KY-11C���ӟ��ͣ�
- ��������R�}�VƬ
- �_��늸�ԇ��25S
- Miki���F݆��ȫ����MSL-T
- �~�S��ʽ���Ѫ��ӋYE8100B
- �~�S݆��܇H005B
- Miki��̫�X�Ͻ���܇��݆��MCS-43L
- �W�Ã|̩늄�݆��EW8603�tɫ(��һ��늳�)
- Miki݆��MOCC-43L
- �X�Ͻ�݆��H008�����
- �Wķ������w��ӋMC-141W
- ����
- ˎƷ���}������
- A
- B
- C
- D
- E
- F
- G
- H
- I
- J
- K
- L
- M
- N
- O
- P
- Q
- R
- S
- T
- U
- V
- W
- X
- Y
- Z
- ���oӛ�
- ���oӛ�
- ���oӛ�
- ���oӛ�
�W(w��ng)վ�YӍ- ˎ����B |
�B�i�T��ֲ� | �˲���Ƹ |
(li��n)ϵ�҂� | �W(w��ng)վ�؈D
�������- ��Ҋ���} | ����(w��)ָ�� | ˎ�W(xu��)����(w��) | ���Ҋ | �Ͷ�V | ���Ʒ���(w��) | ���t(y��)��ˎ | ˎ������
���Ʒ����(w��)- �[���� | �β��� | ��(j��ng)�� | ����� | Ƥ�w�Բ��� | �� �� | �L(f��ng)�����߿� | ��Ѫ�ܿ� | ���� | ��������ˎ
ˎƷ��(d��o)ُ����(w��)- �[����ˎƷ | �����ˎƷ | �β���ˎƷ | �ۿ�ˎƷ |Ƥ�w�Բ���ˎƷ | ��(j��ng)��ˎƷ | �L(f��ng)�����߿�ˎƷ
ˎ���Y�|(zh��)- ��I(y��)���ˠI�I(y��)��(zh��)�� | ˎƷ��(j��ng)�I�S���C | ˎƷ��(j��ng)�I�|(zh��)������Ҏ(gu��)���J(r��n)�C | ʳƷ�l(w��i)���S���C | ��(li��n)�W(w��ng)ˎƷ��Ϣ����(w��)�Y���C
�������- ��Ҋ���} | ����(w��)ָ�� | ˎ�W(xu��)����(w��) | ���Ҋ | �Ͷ�V | ���Ʒ���(w��) | ���t(y��)��ˎ | ˎ������
���Ʒ����(w��)- �[���� | �β��� | ��(j��ng)�� | ����� | Ƥ�w�Բ��� | �� �� | �L(f��ng)�����߿� | ��Ѫ�ܿ� | ���� | ��������ˎ
ˎƷ��(d��o)ُ����(w��)- �[����ˎƷ | �����ˎƷ | �β���ˎƷ | �ۿ�ˎƷ |Ƥ�w�Բ���ˎƷ | ��(j��ng)��ˎƷ | �L(f��ng)�����߿�ˎƷ
ˎ���Y�|(zh��)- ��I(y��)���ˠI�I(y��)��(zh��)�� | ˎƷ��(j��ng)�I�S���C | ˎƷ��(j��ng)�I�|(zh��)������Ҏ(gu��)���J(r��n)�C | ʳƷ�l(w��i)���S���C | ��(li��n)�W(w��ng)ˎƷ��Ϣ����(w��)�Y���C
��(li��n)�W(w��ng)ˎƷ�����Y���C|��(li��n)�W(w��ng)ˎƷ��Ϣ����(w��)�Y���C��|��ICP�䰸̖����ICP09020215
Copyright@2011-2013 �ٝ����ؾW(w��ng)��ˎ��.�ٝ�����ˎ���W(w��ng) m.358cha.cn ���(qu��n)���У����������Й�(qu��n)����
�͑�����(w��)�ᾀ��400-101-6868 Ͷ�V�c���h��020-66843700
����˾���귨������V�|����ʢ�_�Ɏ���(w��)��
����˾���͑��ĺϷ����۷��ɱ��o���κ��ַ�����˾���͑�������κΆ�λ�͂��˱،��ܵ����ɵ�����
Copyright@2011-2013 �ٝ����ؾW(w��ng)��ˎ��.�ٝ�����ˎ���W(w��ng) m.358cha.cn ���(qu��n)���У����������Й�(qu��n)����
�͑�����(w��)�ᾀ��400-101-6868 Ͷ�V�c���h��020-66843700
����˾���귨������V�|����ʢ�_�Ɏ���(w��)��
����˾���͑��ĺϷ����۷��ɱ��o���κ��ַ�����˾���͑�������κΆ�λ�͂��˱،��ܵ����ɵ�����

