
���F(xi��n)�ڵ�λ�ã� �ٝ�����ˎ���W(w��ng)��� >> ˎƷ���} >> �༪��|�������� >> �R���о�
�༪�����R��ˎ�������W�о�
- ��Դ�� �о����փԵ����� ���ߣ��ٝ��ӑB(t��i) �g�[�� �l(f��)���r�g��2007/7/30 11:45:00
����
�˲��փ���Ŀ�����ṩ��Sorafenib�R��ˎ���о������Ŀ��Y���˹�(ji��)���ᵽ���о�������_����ˎ�������M�еĄ����f�����о�������ˎ���W�о������x;�����о�����������Ⱥ���о��Լ����u�rSorafenib�c���������еĿ���ˎ��(li��n)�Õr��ˎЧ�W����ȫ���о��������о��Y������2004��12��31�Ո�����˹�(ji��)߀���������ձ��M�еĄ����f�����о�11497���õ���ˎ�������W�Y�ϣ���2005��4��22�Ո������
�����Y�������M�е�I���о�Ŀ���nj����m�ϵĽoˎ���������_�����L�Ľoˎ�g������Ӱ�ˎ���ȣ���ˌ���N�oˎ�����M�����^�졣���@ЩI���о��еó��İ�ȫ�Ժ�ˎ�������W�Լ�ˎ���Ч�����Y�ϴ�ʹ�ں���_չ���R��II���о��в���400mgÿ�Ճɴε��B�m(x��)�oˎ������
Sorafenib��ˎ�������W�Y���@ʾ��¶���ڻ����g�����^�߲��׃��ϵ��(sh��)�ٷֱȣ�%CV��������36%��91%���c�΄�����ȣ��ڷ�400mgÿ�Ճɴεķ�(w��n)�B(t��i)Cmax�� AUC(0-12)��ֵ�քe������4����4.7�����c�΄�����ȣ��ڷ�400mgÿ�Ճɴεķ�(w��n)�B(t��i)Cmax�� AUC(0-12)��ֵ�քe������4����4.7�����Mʳ��֬���ʳ��Sorafenib�����ս����˼s29%����ˣ��ڷ���Sorafenib�r�����Mʳ�е�֬���ʳ���߽�ʳ��Sorafenib�c��ù��(li��n)����ˎ����ù����HCC���ߵ�AUC��ֵ������21%���в�����@�N������Ƿ�����R�����P�ԡ�
Sorafenib�c�����濵(li��n)����ˎ�������濵�Ļ��Դ��x�a(ch��n)��SN-38�ı�¶���@�����ӣ�67%��120%����SN-38��(j��ng)��UGT1A1�M�д��x���@�N����õ��R�����P��δ����
ˎ������
Sorafenib��ˎ�������W�о����õķ������������о��߆δνoˎ�Ͱ��Y����ÿ�Ճɴνoˎ���ڽ���־Ը���c�е���֬����һ���δη���400mg��Sorafenib�����^�죬��λ�_���ȕr�gtmax�s4-8С�r��ƽ�������Cmax����1.67-2.13mg/L��Ѫ�{���-�r�g�Y���@ʾ��1-2��tmax��������շ壬��ζ�������ѭ�h(hu��n)����(w��n)�B(t��i)ˎ�������W�����Y���߷���SorafenibƬ��400mgÿ�Ճɴκ��M�ж��c�������о�100342��100277��10164��Ѫ�{ˎ�������W�ӱ��ڵ�һ�������һ�νoˎ���ռ�����Sorafenib����λ���Օr�gtmax,ss��3С�r������0.0-24С�r����tmax�������շ���F(xi��n)�ڽoˎ��8-12С�r��/��24С�r���W�ͱ����M�е�I���о��л���ƽ����(w��n)�B(t��i)��Cmax,ss��AUC(0-12),SSֵ�քe��7.7mg/L��64.3mg*hr/L����Ҋ��5-7��
�� 5-7����ԇ�10164��100277��100283��100342�У����Y���߿ڷ��oˎ��ÿ�ՃɴΣ�ÿ�η���400mg Sorafenib��Ѫ�{��Sorafenib������x�a(ch��n)������(w��n)�B(t��i)Cmax�� AUC(0-12)����(sh��)����ƽ��ֵ��%CV��
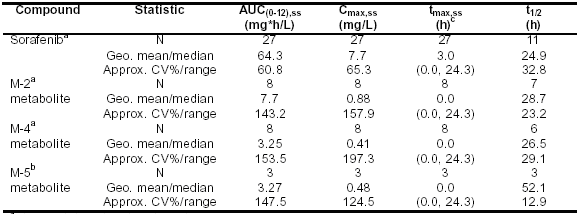
a �oˎ����7���ķ�(w��n)�B(t��i)��(sh��)��(j��)
b �oˎ����21���ķ�(w��n)�B(t��i)��(sh��)��(j��)
c tmax��ƽ��ֵ������
t1/2 ��˥�ڣ��cѪˎ��ȣ��r�g������ĩ��б�����P
Sorafenib��ˎ�������W�Y���@ʾ��¶���ڻ����g�����^�߲��׃��ϵ��(sh��)�ٷֱȣ�%CV��������36%��91%��
��������e
I���R��ԇ��u�r�˶������oˎ��Sorafenib��ѪҺ�е���e���mȻ�����g�����ஔ��IJ�������c�AӋ�Ć�һSorafenib�ĽKĩ��������˥�ھ�ֵ�s��25��48С�r�ĽY����������^ԇ�10164�л���7���21��ı�¶����(sh��)��(j��)���l(f��)�F(xi��n)�oˎ7��֮��Sorafenib�ı�¶���������ӣ����Sorafenib�ı�¶���ڽoˎ��7��֮�Ⱦ��_���˷�(w��n)�B(t��i)���c�΄�����ȣ��ڷ�400mgÿ�Ճɴεķ�(w��n)�B(t��i)Cmax�� AUC(0-12)��ֵ�քe������4����4.7����
�����������ö�
ͨ�^�c����PEG����-80����Һ�����ȣ��u�r50mg SorafenibƬ���������������öȡ��c��Һ����ȣ��c�΄�����ȣ��ڷ�400mgÿ�Ճɴεķ�(w��n)�B(t��i)Cmax�� AUC(0-12)��ֵ�քe������4����4.7����
��ʳ��Ӱ�
�����о���ʳ��Ӱ푣��քe�u�r�˽���־Ը�߷��Æ΄���Sorafenib��ͬ�r�Mʳ��֬���ʳ���е�֬���ʳ���ʳ��B(t��i)�µ�ˎ�������W���Mʳ�е�֬���ʳ��SorafenibƬ�����������ö��c��ʳ��r���ơ��c��ʳ��ȣ��Mʳ��֬���ʳ��Sorafenib�����ս����˼s29%����ˣ��ڷ���Sorafenib�r�����Mʳ�е�֬���ʳ���߽�ʳ��
ÿ��һ���cÿ�Ճɴ�
�քe�u�r��Sorafenib�ԃɷN��ʽ�oˎ��ˎ�������W���΄���200mg��QD����100mgÿ�ՃɴΣ�bid�����ڷ���100mg bid�Ļ��ߣ�24С�r��AUCֵ��200mg QD���߸߃ɱ���ÿ�Ճɴνoˎ���������ö����ӣ����ܚw����Sorafenib��ˮ�еĵ��ܽ�ȣ�����θ�c�����β��ֵ����՜p�١������ÿ��һ����r�£��S���oˎ���������ӣ��������ö��_�����@�ķ�(w��n)�B(t��i)ˮƽ�������҂��x��ÿ�ՃɴεĽoˎ��ʽ���_��������ջ�������ճ̶ȡ�
���������Pϵ
�ڵ���400mg bid������AUC�S������߶�������ͨ���c�������P���c�������������Pϵ��������600mg bid����AUC(0-12),ss��ֵ�c400mg bid��Ȳ�δ�ɱ������ӣ���800mg bid����AUC(0-12),ss��ֵ�c600mg bid���Ҳδ�ɱ������ӡ�
�ֲ�
99.5%��Sorafenib�cѪ�{���Y�ϣ��Y�ϳ̶��c��ȳʾ������P��Sorafenib�ڼtѪ���Ѫ�{�еķֲ�������ȣ�Ѫ�{�cѪҺ֮�Ȟ�1.33��
����
���о�Sorafenib����й�ʹ��x;����ԇ�11195��������λ����־Ը���M�����w���|��ƽ��ԇ�ڷ��΄���[14C] Sorafenib������ԙz�y�l(f��)�F(xi��n)Sorafenib��Ҫ��(j��ng)�S����й���sռ������Կ�����77%����Һ��ռ19%��ԭˎ�ǼS����ȡ���е���Ҫ�ɷ֣��oˎ������50.7%���������M-6�����ữ�����ռ�oˎ������19%����(j��ng)��Һ��й��19%�У���Ҫ�ɷ֣�14.8%����Sorafenib�������ᣨM-7������2.7%��M-2�������ᡪM-8����Һ��δҊSorafenibԭˎ��Ѫ�{������Կ����У�73%��Sorafenib��ʽ�M��ѪҺѭ�h(hu��n)��17%����x�a(ch��n)��M��2������ˎ��W�������ơ�Ѫ�{������������Դ��x�a(ch��n)�������1%��
���Դ��x�a(ch��n)���ˎ�������W
I �ں�II���R��ԇ��о���Sorafenib�Ĵ��x�a(ch��n)��M-2��M-4��M-5�ڰ��Y�����w�ȵ�ˎ�������W���D5��1�����@Щ��(sh��)��(j��)������400mg bid�������������oˎ��M-2�ı�¶���sռ����¶����ԭˎ�ʹ��x�a(ch��n)�ᅡ�ͣ���16%�����x�a(ch��n)��M-2��M-4�ڽoˎ��7���_����(w��n)�B(t��i)����(w��n)�B(t��i)�r���x�a(ch��n)��M-4ռ����¶����8%��M-5ռ6%�����^���x�a(ch��n)��M-5�ڽoˎ���1��͵�21���AUC��Cmaxֵ���l(f��)�F(xi��n)��21��M-5��¶�������Ӹ���Sorafenib��M-2���D5��1����
�w�ȴ��x
�����Sorafenib�ڴ���(73)��Ȯ(74)����(63)�w�ȵ������D��ԇ�Y�����4��5��ʾ����Ŀǰ��ֹ���ڷ��΄���ˎ���ԭˎ�Ǵ���Ȯ����Ѫ�{�е���Ҫ�M�֣��ڷ������AUC������ռ71%��73%�����c����ȣ������ȮѪ�{��M��3��BAY 72-1973���Ӄ�(y��u)�ݵ�λ���քeռ�������AUC������12.1%��15.6%������Ѫ�{��M��2��BAY 67-3472, N-�����a(ch��n)�����Ҫ�Ĵ��x�a(ch��n)�AUC������16.7%�������H�ڴ���Ѫ�{�Йz�y������M��2��0.9%������ȮѪ�{�Єtδ�z����
��4��5���w�������D�����ڷ��΄���[14C]��ӛ��BAY54��9085����(PH-33292)��Ȯ(PH-33063)����(PH-33427)Ѫ�{�е���Ҫ�M��

a ��[14C]Sorafenib�����ɉA��Ӌ
AUC Ѫˎ��ȕr�g��������e
�����w��Sorafenib��Ѫˎ��ȕr�g�������F(xi��n)�ׂ��μ��壬�������ڸ��cѭ�h(hu��n)�����cѭ�h(hu��n)�ij��F(xi��n)�w���������ɷN�^�̣�
�� ˎ����������ữ�a(ch��n)��M��7��(j��ng)đ�ܷ��ڵ��c����������a(ch��n)��ֽ⣬�S��Sorafenib���c�������ա�
�� ˎ��������a(ch��n)��M��2��(j��ng)đ�ܷ��ڵ��c����(j��ng)�Y�c��߀ԭ��Sorafenib���S��Sorafenib���c�������ա�
�H�����w�^�쵽�ɷN��Ҫ�������D��;�����ڴ����Ȯ�tδ�l(f��)�F(xi��n)���cѭ�h(hu��n)�������Ȯ�ڷ�[14C]��ӛ��Sorafenib�ױ������}������Ԏ�ȫ����(j��ng)�S����й�������w�s75%�Ľoˎ������(j��ng)�S����й��ԭˎ���ˣ��oˎ������50.7%���ʹ���(32.3%)�S����ȡ���е���Ҫ�M�֣�����Ȯ�Hռ�oˎ������3.5%�����ˣ������Ȯ�ļS����ȡ���У����ữ�a(ch��n)��M��6�քeռ������Կ�����19.1%��30.0%��62.6%���u�����a(ch��n)��M��3(BAY 72-1973)���DŽ����w�Ȫ��еĴ��x�a(ch��n)��ڴ����Ȯ�ļS���зքe��23.0%��15.0%��
�����w����(j��ng)�I�K����й�ȴ����Ȯ�������@�����˵���Һ��������19%���Йz�y���ɷN���x�a(ch��n)�M��7��Sorafenib���������ữ�a(ch��n)�������14.8%����M��8��M��2���������ữ�a(ch��n)�������2.7%��������δ�l(f��)�F(xi��n)ԭˎ�ɷ֡����^Sorafenib�����w�̈́���������D���l(f��)�F(xi��n)���ɗl���x;�����w����w�ȴ����@����������w����व�N����������(M-2, BAY 67-3472)�ȼ��u��������(M-3, BAY 72-1973)�����@����Sorafenib���������ữ�H�����w�Ⱦ�����Ҫ���x��
�C�������������wSorafenib��(j��ng)�ɗl;���M�������D������CYP3A4����M��2�������Լ���(j��ng)������ȩ���D��øUGT1A9�����������ữ�a(ch��n)��M��7�����ɡ����⣬�����I�KҲ�������������ữ�a(ch��n)��M��7�����ڴ��ڃɗl�����D��;����һ���J��Sorafenib�cCYP3A4���Ƅ���̫���ܰl(f��)��ˎ���g����á����R��ԇ��У�CYP3A4�ď�Ч���Ƅ�ͪ�����cBAY54��9085(li��n)����ˎ����Sorafenib��Ѫˎ��Ȳ�δ�a(ch��n)���κ��@��Ӱ푣�����ܵ�ԭ�����������ữ���x;���l(f��)�]������(75)��
�D 5-1���ڽoˎ21��/ͣˎ7���ԇԇ�10164���У����Y���߷���600 mg bid������Sorafenib���1��7��21�죬Sorafenib������x�a(ch��n)��M-2��M-4��M-5��Ѫˎ���
 �����Y���c���x�a(ch��n)��M-5�İ�˥���^�Lһ�¡��ձ����Y�����ڵ�14��͵�28��Ĕ�(sh��)��(j��)�������oˎ14�����x�a(ch��n)��M-5�_����(w��n)�B(t��i)�����OSorafenib�������N���x�a(ch��n)�M-2��M-4��M-5�������Ƶ��w��ˎ��W���ԣ����Ҹ�ռ��Ѫ�{������ˎ�����P���|AUC������30%���ң���ô�����wѪҺѭ�h(hu��n)��Sorafenib�]����Ҫ�Ļ��Դ��x�a(ch��n)�
�����Y���c���x�a(ch��n)��M-5�İ�˥���^�Lһ�¡��ձ����Y�����ڵ�14��͵�28��Ĕ�(sh��)��(j��)�������oˎ14�����x�a(ch��n)��M-5�_����(w��n)�B(t��i)�����OSorafenib�������N���x�a(ch��n)�M-2��M-4��M-5�������Ƶ��w��ˎ��W���ԣ����Ҹ�ռ��Ѫ�{������ˎ�����P���|AUC������30%���ң���ô�����wѪҺѭ�h(hu��n)��Sorafenib�]����Ҫ�Ļ��Դ��x�a(ch��n)������c�r�g��ه��
������ه��
��I �چ�һ�Ƅ�ԇ��У�������Ѫ���е�Sorafenib������x�a(ch��n)��M-2��M-4��M-5���S����������ߣ������g���w��ஔ����Sorafenib�����������ķN���|��Sorafenib��M-2��M-4��M-5����������ռ�ı���δҊ���@׃����������100mg bid��800mg bid���������ȣ�Sorafenib�Ĵ��x���Є�����ه�ԡ�
�r�g��ه��
�ڎׂ��R��ԇ����о���Sorafenib��ˎ�������W���r�g����ه�ԣ�������ԇ�10164���u�r�˽oˎ���1��7��21���ȫ��Ѫˎ���-�r�g�������@Щ�u�rԇ��Ŀ���ǿ����B�m(x��)�oˎ�r�g��Sorafenib������x�a(ch��n)���ˎ�������W��Ӱ푡�ԇ�l(f��)�F(xi��n)�������oˎ�B�m(x��)7���Sorafenib�� Cmax��AUCֵ�������^�m(x��)���ӡ�ԇ�10164�У����^�oˎ���7���c��21��ı�¶�����l(f��)�F(xi��n)�������oˎ��Sorafenib��M-2��M-4��AUCֵ���������ӡ�
���x�a(ch��n)��M-5���γ��^������˲����ĵ�1�����B�m(x��)�z�yM-5�ĝ�ȡ��Ľoˎ���1�쵽��7��M-5�ı�¶�����ӣ��oˎ��7�쵽21���^�m(x��)���ӣ����ǽoˎ��21�쵽28��ı�¶��������f��M-5�ı�¶���ڶ������oˎ��21����_��(w��n)�B(t��i)ˮƽ�����ձ��M�е�ԇ�10658�У��u�r�˽oˎ���1��14��28���ˎ�������W���l(f��)�F(xi��n)ÿ�սoˎ�ɴκ��14��28��M-5��Cmax�� AUC(0-12)ֵ�����ԇ�10658��ԇ�10164�Ĕ�(sh��)��(j��)�������oˎ���7��10��M-5�ı�¶���_����(w��n)�B(t��i)�����x�a(ch��n)��M-5���γ��^���c���˥���^�L���������
��֮��ԇ�10164��100277��100283��100342��10658�Ĕ�(sh��)��(j��)������Sorafenib��M-2��M-4��M-5�ڽoˎ��14��Ⱦ��_��ˎ�������W��(w��n)�B(t��i)ˮƽ��
�������ص�Ӱ�
�u�r��Sorafenib��ˎ�������W�c�������ص��Pϵ���������ذ������g���Ԅe���w�ء�Ѫ�弡�����Լ��������Ӌ�����ι��ܣ�Child��Pugh A����Child��Pugh B�����Լ�Ѫ��đ�t�صȣ��@һ������u�rʹ����I �ں�II ���R��ԇ���Sorafenib�ί��M���ߵĔ�(sh��)��(j��)���@Щԇ�õ��˻���������Ѫˎ���-�r�g��(sh��)��(j��)��Sorafenib��ˎ�������W�c���g���Ԅe���w�ء�Ѫ�弡����Ѫ��đ�t�؛]�����@���Pϵ����Child��Pugh A����B�����ߣ�Sorafenib��ϵ�y(t��ng)��¶���Ͱ�ȫ�Ԕ�(sh��)��(j��)���ơ����^Sorafenib���ձ��������ߵ�ˎ�������W���l(f��)�F(xi��n)Sorafenib�ڲ�ͬ���ߵı�¶����(sh��)��(j��)��һ���̶����دB��
�C�ϣ�����(j��)�ѵõ���400mg bid��(sh��)��(j��)���o�����(j��)���g���Ԅe���w�أ�������51��116����{���oˎ������
ˎ���gˎ�������W�����
Sorafenib�Ǽ��������Կ���ˎ����ڰ��Y����ͨ����ͬ�rʹ�ö�Nˎ���M���ί������Ժ��б�Ҫ�о��R����Sorafenib�c����ˎ���ˎ�������W����á�ͨ�^�w����x/����/�T����P-gp�D�\�����Ɣ�(sh��)��(j��)������ˎ���g���ڵ���������ԡ����⣬�R�����u�r��ijЩˎ���g������á�����ˎ�{(j��ng)������������ˎ(li��n)��ʹ�ã�����ԇ��u�r��Sorafenib�c��������ˎ���(li��n)����ˎ�������ṩ������ɵ�ԇ(sh��)��(j��)��
�������w����w��ԇ��u�r��Sorafenib�Ĵ��x���c��Sorafenib���w����xԇ�_����I �ں�II ��ԇ��еĴ��x;�����w��ԇ��о���Sorafenib��9�N��CYP�����w��5�N��UDP-����ȩ���D��ø���������á������w�μ����w��ԇ����о���Sorafenib��ø�T�����á������w���u�r��һ���֣����R���|��ƽ��ԇ�Ͱ��Y����I ��ԇ���Ҳ�о���Sorafenib�Ĵ��x�����Y�����R��ԇ��п�����Sorafenib��CYP�����w�x���Ե����Ӱ푡������ֺ�Ҫ���Y�������w����w�ȴ��xԇ�ĽY����
1���w����x��(sh��)��(j��)
�w���˸��K���w��(sh��)��(j��)�@ʾ�� Sorafenib�������x;��I��Ҫ��CYP3A4���������x;��II���c��������Y�ϣ���Ҫ��UGT1A9�������x�a(ch��n)��M-1��M-5���������x�a(ch��n)�M-6���ữ���x�a(ch��n)�M-7��M-8�քe��Sorafenib��M-2���������ữ�a(ch��n)�
�w��ԇ��u�r��Sorafenib�ױ������}��9�N��CYP�����w���������á�δҊSorafenib��CYP1A2��CYP2A6��CYP2E1���Ԯa(ch��n)���������ã�Sorafenib��CYP2C19��CYP2D6��CYP3A4�����ж��������ã�Kiֵ�քe��17 M��22 M��29 M��CYP2B6��CYP2C8��CYP2C9�Ļ����ܵ�Sorafenib�����������ƣ�Kiֵ�քe��6 M��1��2 M��7��8 M��Sorafenib�����x;��II���Y�Ϸ�����øUGT1A1��UGT1A9Ҳ�����������ã�Kiֵ��1��2 M����400mg bid������Sorafenib�ķ�(w��n)�B(t��i)Ѫˎ���Cmax��ֵ��16.6 M��7.7 mg/L��������Sorafenib��Cmaxֵ�����īI����Ķ���(sh��)Kiֵ�����Sorafenib���܌��@ЩCYPs������w�ȴ��x���Н����������á�
�R��ԇ��^�m(x��)�u�r���ܵ�Sorafenib���Ƶ�CYPͨ�����R�������P�ԡ����Sorafenibͨ�^�ɗlƽ�е�;���M�д��x������Sorafenib�c��һ;������ˎ��(li��n)����ˎ�r����¶���@�����ӵĿ������^С��ͪ�����R������ԇ�Y���C�����@һ���O��ԇ�δҊ���@������á�ͬ�ӣ�Ҳ�]�аl(f��)�F(xi��n)CYP1A2��CYP3A4���T�����ã��f���R���ϰl(f��)��ø�T���Ŀ����Ժ�С���cԇ�10926�ĽY��һ�¡�
2���w��P-�ǵ��ף�P-gp�����Ɣ�(sh��)��(j��)
Caco-2������ͨ���u�r����Sorafenib�ǸߝB�����Caco-2�������_P-gp��Sorafenib��Caco-2�����ׂ�픲����ُ�픲��ׂ��D�\�ĝB���Ȟ�2.9��4.7�����OSorafenib�иߝB�ԣ��AӋ�����B������Ӱ��������w�����w���ա�
�w��ԇ��@ʾSorafenib�ױ������}�����wP-gp�B���þ����ஔ����������á�����̽ᘻ��������߶������p���_Ī�B����IC50ֵ�քe��0.84µM��1.24µM�����R��ԇ��У���400 mg bid������Sorafenib��Ѫˎ��ȸ߳�����IC50ֵ�ױ�֮�࣬����Sorafenib���܌���(j��ng)P-gp;���D�\��ˎ����Н����������á��p���_Ī�cSorafenib(102)һ�ӌ�P-gp�����������á������p���_Ī��������ظ�����¶���@�����ӣ����Sorafenib�cP-gp����(li��n)����ˎ�r���R���Ͽ��ܲ����l(f��)�����@������á��c�˽YՓ��һ�µ����Ј��ָ����P-gp���cˎ���������P������Á��f���ܲ�̫��Ҫ����103��
3��CYP3A���Ƅ�ͪ����Sorafenibˎ�������W��Ӱ�
�w��ԇ�l(f��)�F(xi��n)Sorafenib���������x;��I��Ҫ��(j��ng)��CYP3A4�������ںܶ���������|ͨ�^CYP3A4�M�д��x���S��ˎ���g�������Ҳ�cCYP3A4���P�������҂�������CYP3A4�ď������Ƅ�ͪ����Sorafenib��Ӱ푡�ԓԇ��У�Sorafenib�oˎ������50 mg��ͬ�rÿ�����ͪ����400 mg���m(x��)7�졣�Y������ͪ����δ����Sorafenib�ı�¶��������Ѫˎ��ȣ�����5��8����
�� 5-8��Ѫ�{��Sorafenib�� AUC��Cmax����(sh��)���Լ��@Щ����(sh��)���cͪ����(li��n)��/δ�cͪ����(li��n)�Õr�ı�ֵ(����Сƽ����ֵ) (ԇ� 10927)
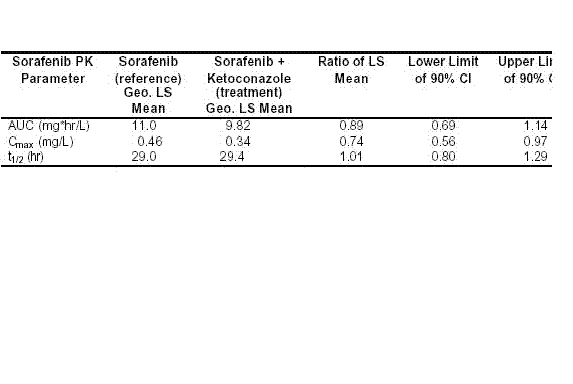 AUC = Ѫˎ��ȣ��r�g��������e
AUC = Ѫˎ��ȣ��r�g��������eCI = ���Ņ^(q��)�g
Cmax = ���Ѫˎ���
PK �C ˎ�������W
Geo. LS mean = ����Сƽ����ֵ
t1/2 = ��˥�ڣ��cѪˎ��ȣ��r�g����ĩ��б�����P��
�cͪ����(li��n)����ˎ�r��Sorafenib�ı�¶��δҊ���ӣ���˿����Ɣཛ(j��ng)��CYP3A4�����������x;��I��Sorafenib�Ĵ��x�в�̫��Ҫ���@Щ��(sh��)��(j��)�Y��III��ԇԇ�11213����(sh��)��(j��)��ԓԇ���41λSorafenib�ί��M�����c19λ��ο���M����ͬ�r����CYP3A4���Ƅ����Y�������R����Sorafenib�cCYP3A4��������Ƅ�(li��n)����ˎ�ǰ�ȫ�ġ�
ͪ������P-gp�D�\�ď������Ƅ��������R��ԇ���δҊͪ����Sorafenib��ˎ�������W�a(ch��n)���κ�Ӱ푣����Կ��Ɣ�P-gp�D�\����Ѫ�{��Sorafenib��ˎ�������W���f������Ҫ��
�c��������ˎ��(li��n)�õ������ԇ�
δҊSorafenib�c�������I��Wɳ���K�l(f��)��һ�µĻ������x��ˎ�������W����ã�����Sorafenib�����c�������I��Wɳ���K(li��n)����ˎ��
Sorafenib�c��ù��(li��n)����ˎ����ù����HCC���ߵ�AUC��ֵ������21%���в�����@�N������Ƿ�����R�����P�ԡ�
Sorafenib�c�����濵(li��n)����ˎ�������濵�Ļ��Դ��x�a(ch��n)��SN-38�ı�¶���@�����ӣ�67%��120%����SN-38��(j��ng)��UGT1A1�M�д��x���@�N����õ��R�����P��δ����
4���w���u�r����ɫ��P450��Sorafenib����������
���ڰ��Y�����M�е��R��ԇԇ�10926���У������L�ڷ���Sorafenib�������һCYP�����w���������Ƿ�����R�����P�ԣ�������Sorafenib ��CYP2C19(�W������)�� CYP2D6(����ɳ��)��CYP3A4 (���_���)̽ᘵ����Ӱ푡�
���Y���߶�η���400 mg bid������Sorafenib��CYP3A4(���_���)��CYP2D6 ������ɳ�ң���CYP2C19(�W������)̽ᘵ����ˎ�������W��δ�l(f��)���@��׃��������R����Sorafenib�ȷ�CYP2C19��CYP2D6��CYP3A4ͬ��ø�����Ƅ���Ҳ���������T������Sorafenib�����@�����ӻ�p�ٽ�(j��ng)���@Щ;�����x�Ļ�����ı�¶����
�w��ԇ���Sorafenib������CYP2C9��Kiֵ��7��8µM������S-�A������Ҫͨ�^CYP2C9;�����x����ԇ�11213�У�ͨ�^PT-INR�����g�әz�ySorafenib���A������x��Ӱ푡�PT-INR����(sh��)��Sorafenib�M�c��ο���MδҊ����f�����w��Sorafenib���ܲ���CYP2C9�����Ƅ���
TAG���༪��
���PˎƷ




�W(w��ng)վ�YӍ- ˎ����B |
�B�i�T��ֲ� | �˲���Ƹ |
(li��n)ϵ�҂� | �W(w��ng)վ�؈D
�������- ��Ҋ���} | ����ָ�� | ˎ�W���� | ���Ҋ | �Ͷ�V | ���Ʒ��� | ���t(y��)��ˎ | ˎ������
���Ʒ����- �[���� | �β��� | ��(j��ng)�� | ����� | Ƥ�w�Բ��� | �� �� | �L�����߿� | ��Ѫ�ܿ� | ���� | ��������ˎ
ˎƷ����- �[����ˎƷ | �����ˎƷ | �β���ˎƷ | �ۿ�ˎƷ |Ƥ�w�Բ���ˎƷ | ��(j��ng)��ˎƷ | �L�����߿�ˎƷ
�ٝ�ˎ���Y�|- ��I(y��)���ˠI�I(y��)��(zh��)�� | ��(li��n)�W(w��ng)ˎƷ��Ϣ�����Y���C
�������- ��Ҋ���} | ����ָ�� | ˎ�W���� | ���Ҋ | �Ͷ�V | ���Ʒ��� | ���t(y��)��ˎ | ˎ������
���Ʒ����- �[���� | �β��� | ��(j��ng)�� | ����� | Ƥ�w�Բ��� | �� �� | �L�����߿� | ��Ѫ�ܿ� | ���� | ��������ˎ
ˎƷ����- �[����ˎƷ | �����ˎƷ | �β���ˎƷ | �ۿ�ˎƷ |Ƥ�w�Բ���ˎƷ | ��(j��ng)��ˎƷ | �L�����߿�ˎƷ
�ٝ�ˎ���Y�|- ��I(y��)���ˠI�I(y��)��(zh��)�� | ��(li��n)�W(w��ng)ˎƷ��Ϣ�����Y���C
��(li��n)�W(w��ng)ˎƷ�����Y���C|��(li��n)�W(w��ng)ˎƷ��Ϣ�����Y���C��|��ICP�䰸̖����ICP��17064671̖-3
Copyright@2013-2021 �ٝ����ؾW(w��ng)��ˎ��.�ٝ�����ˎ���W(w��ng) m.358cha.cn ������У����������Й�����
�͑����՟ᾀ��400-101-6868 Ͷ�V�c���h��020-29859339
Copyright@2013-2021 �ٝ����ؾW(w��ng)��ˎ��.�ٝ�����ˎ���W(w��ng) m.358cha.cn ������У����������Й�����
�͑����՟ᾀ��400-101-6868 Ͷ�V�c���h��020-29859339

